| HapMap五周年回顾 |
| 2011-01-12 | 作者: 【关闭】 |
|
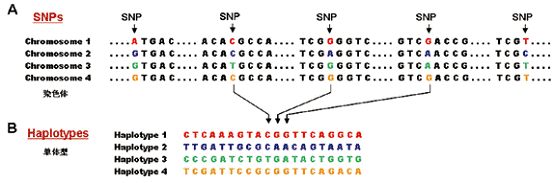
作者简介:曾长青,中国科学院北京基因组所研究员,博士生导师。CUSBEA奖学金、百人计划、杰出青年基金、首批新世纪百千万人才工程国家级人选获得者。作为“十五”重大攻关项目课题组长、国际HapMap计划Steering Committee Member和 “中华单体型图协作组”召集人,负责HapMap“中国卷”的实施。主要从事疾病相关基因定位、群体遗传学和基因组多态研究,部分成果在Nature,Nature Genetics, PNAS等杂志发表。 今年10月,是人类基因组国际HapMap计划启动8周年和这一重大国际合作的主要任务完成5周年。几年间,全球范围的基因组特别是人类基因组的研究,已经成为生命科学的最前沿学科,HapMap也成为应用最为广泛、深入并不断完善更新的人类最大数据库。本文回顾性介绍HapMap计划及其近年来在基因组学和人类健康领域的重要作用。 1、人类基因组的HapMap和国际HapMap计划 (1)何谓HapMap HapMap是Haplotype Map 的简称,Haplo意为单一,在基因组中专指来自父母的一对染色体中的一条。Haplotype就是单条染色体中的一段,译作单体型(有人译作单倍型),是描述遗传差异的一种主要方式。DNA作为遗传物质,不但编码了物种间的差异,物种内不同个体之间的差异也含在其中,均表现为基因组之间的DNA序列差异,也就是基因组的多态性上。 DNA由四种核苷酸单个连接而成,基因组最常见的多态就是单核苷酸多态(single nucleotide polymorphism),简写为SNP或SNPs(复数),指在群体中染色体的某一位点上由不同的核苷酸构成(图1)。
图1 单核苷酸多态位点SNP和单体型。A:在来自4个个体的一段6kb序列上,大部分核苷酸相同但有5处显示不同(彩色)即SNPs。B:这段6kb区域的20个临近SNPs组成单体型(Haplotypes)。摘自Nature. 426:189-193。 目前发现的人类基因组中的SNP位点已经超过一千万。在人群中,染色体上每一二百个核苷酸就有一个SNP位点。单体型描述的是一段单条染色体上的序列差异,就是由SNP位点的顺序排列组成。因此也可以说单体型是分别来自父母的单条染色体上SNP的分布和传递模式。根据邻近SNP的连锁特性(即连锁不平衡),单体型上的多个SNPs还可以由少数几个tag即标签SNP代表。Haplotype Map是单体型图谱,就是全基因组上所有DNA序列的SNP分布和人群频率、标签SNPs、连锁性质与规律等。 (2)HapMap的内容 HapMap的内容是一个巨大的“人类遗传用表”。遗传学研究的本质在于解析变异及其传递规律。对于人类基因组中的千百万个SNPs来说,尽管很多SNPs对于表现型可能并没有贡献,个体的性状差异,也就是任意两个人之间的遗传差别,就蕴藏在这些SNPs和单体型之中。如果说人类基因组测序计划打开了我们自身的遗传密码这本天书,那么地球上每个人的天书都是一种版本,HapMap揭示的就是不同天书版本之间的差别与规律。从个体的基因组测序到全基因组SNPs在人群中的分布和单体型的构建,HapMap计划标志着群体基因组学研究的开始,旨在通过对于海量SNPs的人群检测及其计算分析,构建全人类的遗传用表,从而研究者可以从中查到基因组中各种位置的SNP以及在主要人群中的组成、频率和单体型与标签SNP情况。 (3)HapMap的重要价值 HapMap的重要价值在于揭示复杂性疾病的遗传因素。复杂性疾病是相对于单基因病(如血友病等)而言,也是常见疾病,如高血压、肿瘤、精神性疾病、糖尿病等。其发生往往是遗传和环境因素共同作用的结果,遗传因素涉及多个基因和分子通路,而不表现为单基因的孟德尔遗传。因此罹患这类疾病的易感性,包括对不同环境或药物的敏感性等,都隐匿在多个基因的微效变异即SNP和单体型及其组合中,并且有很大的个体差异。因此,揭示这类疾病的遗传模式需要大量的群体样本和发现与检测极多的SNPs进行关联分析,这在HapMap构建之前几乎是不可能的。国际单体型图计划(TheInternational HapMap Project)的主要目的就是构建不同人群的高密度SNP图谱,通过分析计算确立单体型及其中SNPs的连锁性质和标签SNPs,从而使研究人员可以根据这一巨大的遗传图表和所揭示的人类群体的分子遗传机制,为发现复杂性疾病的相关易感基因确定研究方案和选择需要进行分析的标签SNPs。 (4)HapMap计划的启动和主要任务的完成 国际单体型图计划经过3年的筹划,于2002年10月29日在华盛顿召开由加拿大、中国、日本、尼日利亚、英国和美国6个参加国代表出席的第一次战略会议。会后协作组(Consortium)召开新闻发布会向全球宣布这一计划的正式启动。HapMap计划参加国中加、中、日、英和美国分别承担全基因组10%,10%,25%,24%和31%的SNP分型任务。尼日利亚提供非裔样品,中国和日本共同提供亚裔样品,美国提供了欧裔样品。第一次战略会议上决定了HapMap两阶段的战略。第一阶段是针对非、亚、欧裔的270个DNA样品在全基因组范围以平均每5 000个核苷酸(5 kb)一个SNP的密度进行大规模SNP分型鉴定,构建5 kb单体型图。第二阶段是将HapMap的分型密度增至2kb左右。HapMap计划的运作由各国代表组成的Steering Committee及其下属的若干工作小组负责执行实施。 3年后的2005年10月26日,由国际协作组总负责人,现任美国NIH负责人Francis Collins向全世界宣布了一个拥有数亿数据的人类基因组单体型图的成功构建,以及一个更精细的遗传图谱即将完成(图2)。
图2 2005年10月26日美国盐湖城HapMap新闻发布会场,国际协作组负责人,时任美国国立人类基因组研究所所长FrancisCollins代表所有参加国和国际协作组宣布人类基因组单体型图的圆满完成。 各中心的巨大努力使分型密度比预计提高了近40%,共针对一百多万SNPs构建了密度约3.6kb的数据库和HapMap。美国卫生和公共服务部部长Mike Leavitt在会上评价和描述了HapMap计划的意义和前景。新闻发布会上还发行了载有HapMap及其长文“人类基因组的单体型图谱”的Nature杂志。HapMap这一任务的完成,也标志着这个巨大项目的“中国卷”的完成。中国科学家为这一计划做出了10%的贡献。 (5)HapMap计划的后期工作和进展 HapMap计划的第一阶段任务完成后,国际协作组委托Perlegen Sciences 完成第二阶段扩大SNP分型密度的任务。2007年10月18日,国际协作组在Nature上发表了根据第二阶段数据构建的人类基因组的第二代HapMap。至HapMap 二期共发现了超过一千万的人类基因组的SNPs,完成了约310万SNPs(≥5%)在270个样品中的分型反应。这些SNPs约占预测的遗传变异的25%~35%,并使第二代HapMap的分辨率达到平均不到1kb一个SNP,比预定计划超过100%,准确度达到99.8%。 为使HapMap具有更大参考性,在前两个阶段近10亿SNP数据的基础上,一个新的人群分析HapMap 3启动并于2010年9月在Nature上发表了新的海量数据。与前两期内容不同的是,HapMap 3旨在大量扩充人群样本和发现低频率SNPs。共160万的常见SNPs在来自全球11个人群的1 184个体中进行了分型反应,使HapMap具有更广泛的代表性。同时,还在其中692样品中进行了1Mb区域(10kb×10)的重测序,以发现新的低频率SNPs。显然,随着更多数据的产生,人类基因组的HapMap将不断更新使之涵盖更多人群特异数据和具有更为精细的分辨率。 2、重新了解的基因和染色体——HapMap对于人类基因组结构的重大贡献 (1)基因含义的修订 早在2005年之初,HapMap尚未完成之时,Science就预测这一计划的完成将是生命科学取得的最重大进展之一;至当年底HapMap又被两院院士评为世界10大科技进展中的第5位。的确,HapMap计划的结果与应用及其由此引出的更多基因组规模的深入研究带来了遗传学和基因组学的全面知识更新,甚至包括现代分子生物学概念中的基因与染色体的结构。作为HapMap的卫星项目开始的ENCODE 计划(Encyclopedia Of DNAElements,DNA元件百科全书)于2007年宣告完成。这一扩展到涉及11个国家80家机构参加的研究通过整合计算测序数据和实验分析,鉴定出人类基因组中所有的功能组分,包括编码基因、非编码基因、调控区域、染色体结构维持和调节等所有类型DNA元件的分布和组织方式。合作组同时在Nature和Genome Research上发表了29篇论文,详尽描述了在所分析的1%人类基因组区域内最为完整的生理功能元件,还对很多传统的分子生物学定义做出颠覆性扩展或修订。例如,microRNA,非编码RNA等的普遍存在使“一个基因一个酶”,“中心法则”等都不再是生命科学的金科玉律。这些革命性进展为进一步认识基因组的功能蓝图开辟了道路,对疾病的遗传研究产生了重大影响。 (2)全新的结构差异和拷贝数变异概念 虽然HapMap计划的目标是对于单核苷酸多态位点的模式分析,一个未曾预见的重大成果是通过对于单体型的分析所发现的基因组中同样广泛存在的结构差异(structural variation,SV),包括DNA序列的插入、删除、倒位、易位等。这些结构差异大小很不均一,可在数十个至数百万核苷酸之间。HapMap 揭示出结构变异是基因组中一种常见但由于经典检测技术的缺陷而知之甚少的遗传多态性。由于很多结构多态可在HapMap这一高密度差异图谱上留下“印记”,近年来随着对HapMap的分析利用使人类对于自身染色体的结构多态性产生了全新的认识。仅2006年就有3个小组从不同角度构建了人类基因组的缺失图谱。随着对DNA片段的插入和删除愈来愈多的发现,拷贝数变异(copy numbervariation, CNN)成为描述这类遗传差异的最新名词。这类新发现的遗传变异覆盖了人类基因组大约20%的区域,估计10%~20%的可“调节”基因活性的遗传变异是CNV。同SNP一样, CNV可导致先天性疾病并与多种复杂性疾病的发生有关。HapMap的完成不但导致了人类基因组拷贝数目多态性计划(The CNV Project)的启动,也为高通量筛查CNV 提供了全新的研究策略和实验手段。需要指出的是,拷贝数变异多发生在重复序列,目前对于“断点”位置的精细确定极具技术挑战性。因此一些研究曾导致CNV在基因组中的覆盖度达40%~60%的过高估计。准确定位CNV有待于更先进手段如单分子测序等的发展。 3、HapMap计划对于基因组科学和系统生物学的极大推动 如果说人类基因组计划奠定了基因组学的基础,HapMap计划则开启了群体基因组学的时代,并且在很大程度上参与催生了系统生物学的发展。HapMap提供了全基因组SNPs 的群体分布图谱并揭示了人群内的遗传结构,不但构建了“群体基因组学(populationgenomics)” 基础还建立了一种新的研究策略。这些使自然选择和人群演化成为近年来基因组学研究的又一热点。基于此,近年来还获得了若干人类阳性自然选择图谱和数据库。尤其值得一提的是,2010年我国多个小组通过独立或合作研究,数月间先后在Science,PNAS,Mol. Biol. Evol.刊登多篇文章阐述藏族对于高原低氧适应性的分析结果,所有研究均采用HapMap的汉族数据作为对照进行比较。这些成果不但为深入的群体遗传学研究和疾病的遗传因素及基因功能分析提供了新的视野,还大大加深了人类对于环境的适应过程和人群表型分化的认识。 HapMap计划不但带来更多全基因组规模的大型研究,还大大推动了对多个物种的系统基因组学研究。如上述ENCODE 计划完成之后,一个更大规模的多物种ENCODE项目又开展起来,以解析ENCODE保守区域在不同物种中的细节,进一步了解物种演化过程中的基因组系统变化。利用HapMap数据进行的基因表达研究,还为分子通路和网络研究奠定了数据基础。正是这些研究的汇集和整合,促进了系统生物学的产生和深入开展。 4、应用遗传用表解析疾病原因 人类遗传用表HapMap已经广泛用于疾病研究。HapMap计划的立项目的是为复杂性疾病的基因定位研究提供基础数据、研究策略和先进技术。在此之前,复杂性疾病的遗传研究始终缺乏有效方案。主要原因在于这些疾病不但是遗传和环境因素共同作用的结果,并且其遗传因子涉及多个基因和多条基因产物互作的分子网络或代谢通路。在这类疾病的遗传因子中每个基因的变异对于疾病的贡献并不显著,但其某种未知整合却成为个体罹患疾病的风险。同时,这些不同的易感基因或位点在疾病中的作用有很大的个体差异。因此,如果在群体规模将所有的SNPs进行筛查以确定疾病的相关基因,这在耗资上是无法做到的。HapMap计划应用基因组学“单体型板块”、“标签SNP”等原理,一方面描述了个体差异位点的分布和传递规律,另一方面构建了以人群中遗传差异的传递模式为主要内容的图谱。其中无冗余的分型SNPs超过310万,连同用于质控和低频率SNP位点,整个HapMap计划在270个样品中进行了分型的SNPs达到630多万,超过所估计存在的人类SNPs数量的一半。这些供全球免费下载的数十亿份数据,为各种规模的病例-对照关联分析提供了极为重要的工具和数据,使大规模的对于复杂性疾病的关联分析成为可行的易感基因定位手段。对于较为传统的使用家系样品进行的单基因病连锁分析,HapMap的海量数据使高密度SNPs成为比传统的微卫星标记有更高的解析度和准确率的遗传标记,并使连锁与关联分析的联合使用成为更为有效的定位方法。 从HapMap计划对外发放数据,就揭开了通过SNP分型进行病例-对照的关联分析的序幕。HapMap的完成使候选基因或全基因组的病例-对照关联分析在全球范围得到普及,并且促进了几个“超大型”的复杂性疾病遗传因素分析计划的立项和实施,包括GAIN(geneticAssociation Information Network,遗传关联信息网络)、POPRES(POPulation REference Sample,群体参照样本)、WTCCC(The Welcome Trust Case-Control Consortium,Welcome基金会病例对照协作组)和多个针对某一疾病如高血压等的大型联合研究。其共同特点是采用包含标签SNPs的全基因组芯片,进行GWA(genome-wideassociation)研究,又称GWAS(GWA studies),即不考虑先验知识直接通过对大样品量(上千份)进行整个基因组的关联分析研究。例如,2007年11月WTCCC报告了对超过16000个样本所进行的针对双向情感障碍等7种常见复杂性疾病的GWAS结果,在克罗恩病(Crohn’s disease)等5个疾病中发现至少一个易感基因信号。GAIN 计划则为注意力分散/多动症等6种复杂性疾病的GWAS研究提供支持,寻找易感基因和新的诊断方法。HapMap作为人类遗传用表已经广泛用于疾病相关基因定位研究。 复杂性疾病相关基因定位研究是长期探索和缓慢积累的过程。然而,大量全球性的大规模GWAS研究亦逐渐暴露出许多问题和局限,包括部分大型研究没有发现显著信号,不同人群结果重复性差,无法解释疾病相关信号的生物学意义,易感性对于疾病的真正贡献缺乏评估和与指导临床相脱节等。这些实际上显示出揭示多基因常见疾病遗传机制的复杂性和难度,也反映了一些初期对于使用现有方法一举解决复杂性疾病遗传机理的期待可能过于乐观简单。对于一种涉及多种分子通路的复杂性疾病来说,发现其所有的遗传因素也许如同完成一幅复杂的拼图(jigsaw),需要大量元件的积累和逐渐拼接。以现有基因组学知识考虑和综合分析GWAS结果,人群的遗传背景差异和部分易感性位点是较低频SNP可能是GWAS信号不显著和人群重复性差的重要原因。此外,样品收集过程中临床标准的取舍、分类、控制等方面的差异,也可能降低重复试验的分辨率。对此,将千人基因组等项目发现的低频率SNPs也整合至GWAS研究中,以及扩大样品量或进行若干个类似GWAS的联合分析即Meta analysis,将使发现阳性信号的可能性大为提高。最近,一个由超过100个研究中心参加的糖尿病相关国际协作组对总共8 000多个病人和近40 000对照进行荟萃分析,发现了12个与胰岛素分泌相关的II型糖尿病的易感基因和位点。此外,随着越来越多的远距离调控因子及非编码RNA的发现,非基因区的信号也将获得越来越多的解释。毋庸置疑,与基因组学及其先进技术的迅速更新相比,在HapMap基础上以关联分析为主的大规模复杂性疾病易感基因的定位研究无疑将是一个长期探索和缓慢积累才能最终全面整合结果的过程。 5、HapMap计划对于中国基因组科学的重大推动 如同人类基因组计划,HapMap计划的完成是一个里程碑式的成就,从研究策略到分析与推算的多种手段和算法,均代表了最先进的科研成果和进展,具有极高应用性和创新性。HapMap“中国卷”为构建占人类基因组10%的3号、21号染色体和8号染色体短臂的单体型图以及提供一半的亚洲样品。主要内容于2002年纳入国家“十五”科技攻关计划,其中2.5%的任务由香港创新科技署和香港大学教育资助委员会联合资助的香港小组完成。HapMap中国卷10%的任务量对于承担团队是极为严峻的考验和挑战。HapMap其他成员都是拥有高通量SNP分型体系和经验丰富的一流中心,只有中国团队从购买安装设备开始,各方面从一开始就落后他人一年。特别是,北京课题组资金严重不足。双重巨大压力下团队背水一战,超负荷追赶,采取一系列措施提高反应能力和降低成本,节省一切可能经费,最终在精诚合作和相互帮助下,中国团队完成了超过3 500万的SNP分型反应,保质保量地完成了中国卷任务并成为最早完成补洞的国家。 从人类基因组计划的1%到HapMap计划的10%,中国同发达国家一起参加了揭示人类遗传机制全貌的两个宏伟计划,也是其中唯一产生数据的发展中国家。1%项目的参与使我国在基因组学这一学科领域得到迅速起跑,HapMap10%的贡献则标示着中国科学在基因组学这一新兴学科中的飞跃发展。这一历史过程不但显示了我国在基因组学的巨大进步以及国民和国力对于这一人类最大公益项目的支持,还体现了中国科学家抓住历史机遇、把握学科前沿进展,从落后领域冲天而起的拼搏与成功。中国的参与发展训练了研究队伍,并极大促进了基因组学在中国的开展。在中华民族的遗传多态和重大疾病的基因组研究方面,多项重要研究成果和高水平论文每年都在递增产生。我国在这一领域已经获得大量世界领先成果并将以更大的生命力持续发展下去。 6、基因组科学带动的先进技术手段的发展 基因组科学发展的一个重要特点就是与先进技术手段极其紧密的相互结合和促进,正是这种密切关系产生了科学与技术共同的迅猛发展。同早期测序技术落后的情况不同,多种比较成熟的SNP分型技术在HapMap计划的实施之初已经发展起来,从而保证了HapMap计划的3年圆满完成。HapMap计划不但有力推动了SNP技术的快速发展,还大大促进了多种基因组学技术的研发。特别是多种密度的全基因组SNP芯片为病例-对照研究提供的有效工具,几乎被所有大型疾病相关研究采用。同时,在SNP杂交芯片基础上,多种全基因组规模的其他分子检测芯片陆续开发出来,包括定量检测转录本表达、microRNA、DNA甲基化等检测芯片,已在人和多种模式生物中获得大量应用。通过基于SNP或微列阵的比较基因组杂交(comparative genomic hybridization,Array-CGH),则成为发现和研究拷贝数目差异的重要手段。 值得一提的是用于SNP分型的高通量芯片杂交技术还催生了应用微型“霰弹法”测序策略的二代测序技术的研发,近两三年来以高通量为特点的新一代测序仪已经成熟并迅速占领了市场。无论在研究策略和手段,还是分析内容的种类和深度上,二代测序仪的普及都带来了基因组学研究的革命性思路和进展。如群体基因组规模的测序项目千人基因组计划的前期工作(Pilot project)已由二代测序手段完成,包括对两个三体家系的深度测序(平均42X),159个HapMap样品的低度测序(2-6X)和对697个体的部分外显子测序。美国NIH启动的肿瘤基因组计划(The Cancer Genome Atlas,TCGA)目前正在以胶质细胞瘤、肺癌和卵巢癌为先导,通过二代测序进行肿瘤基因组的分析。此外,在SNP杂交芯片的基础上与二代测序同时发展起来的还有通过分子探针进行基因组上目标序列捕获的技术。根据研究需要的定制靶点探针和针对全基因组编码部分的商品化外显子探针将目标序列从整个基因组上特异性捕获下来,与二代测序联用,已经成为一种新的研究方法,极大促进了相关基因定位、医学重测序等复杂性疾病研究。 7、公益事业的光辉典范 作为重大国际性公益事业,HapMap的成功合作和完成,再一次打破基因专利对于人类健康事业的挑战。自人类基因组测序的工作框架图发表,研究SNP和基因组多态性便成为新的研究热点。特别是,人类基因组的SNP图谱是通向确定复杂性疾病易感基因与位点的钥匙,导致又一次出现了多个私营集团因基因专利的巨大商机而多方位兴起的全基因组规模研究热潮,再次使人类基因组计划所提出的数据共享原则受到严重挑战。由此,国际协作组提出了人类基因组研究的第二个战略任务,以多国合作形式共同构建单体型图的国际HapMap计划应运而生。为了与可能的基因或SNP专利赶超时间,协作组特别采取了根据项目进度随时公布数据的策略。在HapMap网站上将所有阶段性数据即时发出供全球研究人员无偿使用(www.hapmap.org)。最终HapMap的所有数据,包括低频和罕见SNPs、分型技术、算法与结果等,全部无偿公布,供全球其他研究所用。 这些举措使人们再一次解除了对大规模疾病相关基因和位点的专利的忧虑。如同测序计划的全人类共享运作与私营企业试图抢先基因专利的激烈竞争的一个更大规模的重演,最终受政府支持的公益项目以其无法超越的规模和速度迫使多个启动更早的企业相关项目,特别是针对复杂性疾病的计划放弃对于SNPs的专利尝试。不止一个企业有偿或无偿地将数据纳入到HapMap计划中,甚至从竞争转为重要参加者。最终一个高密度的HapMap和目前拥有数十亿数据的人类基因组多态数据库成为全人类共有的宝贵财富。 还在HapMap计划酝酿之时,少数资深学者对于这一耗资甚大的计划所构建的HapMap能否为后续的疾病相关基因研究提供重要数据怀有疑问并影响到一些我国学者。同时还存在是否“值得”耗资参加这样的公益项目的疑问。对此,自2004年开始显露的全球范围对于HapMap结果的大量应用已经充分证明了HapMap数据对于遗传多态和基因组研究的不可替代的重要意义,HapMap的奠基用对于后续开展的系列全基因组规模研究的巨大作用实际上大大超出预料。而中国作为参加国之一,目前在基因组学方面获得的瞩目成就亦早已证明益莫大焉。HapMap计划已经将基因组学研究引向新的阶段,对人类健康产生着深远影响。一个全面揭示所有复杂性疾病的相关基因和人类遗传机制的时代正在来临。 本文发表于:《科学新闻》中国生物研究热点论文特约稿——Vol. 5 No. 6 2010 |